Número Atual: Janeiro-Fevereiro 2016 - Volume 4 - Número 1
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

RELATO DE CASO
Síndrome de ataxia telangiectasia sem ataxia: relato de caso
Ataxia-telangiectasia syndrome without ataxia: a case report
María Claudia Ortega López, MD; Agatha León Quintero, MD
DOI: 10.5935/2318-5015.20160006
Fundación Universitaria de Ciencias de la Salud-Hospital Universitario Infantil de San José, Bogotá, Colombia
Endereço para correspondência:
Maria Claudia Ortega Lopez
E-mail: mcol19@yahoo.com
Submetido em 16/02/2015
Aceito em 5/12/2016
Nao foram declarados conflitos de interesse associados à publicaçao deste artigo.
RESUMO
A síndrome de ataxia telangiectasia (AT) é uma síndrome complexa, com herança autossômica recessiva, de baixa incidência, que envolve múltiplos órgaos. É caracterizada por distúrbios neurológicos, ataxia cerebelar progressiva, imunodeficiência variável com susceptibilidade aumentada a infecçoes sinopulmonares, hipersensibilidade aos raios-X, telangiectasias óculo-cutâneas, predisposiçao para tumores, instabilidade cromossômica e altos níveis de alfa-fetoproteína. A maior parte das mutaçoes identificadas no gene AT mutado (gene ATM) causam truncamento da quinase de ATM, responsável pela reparaçao de DNA e regulaçao do ciclo celular. Descrevemos paciente do sexo feminino de 8 anos de idade, de pais nao consanguíneos, com antecedentes de infecçoes recorrentes, telangiectasia ocular, apraxia ocular, deficit de crescimento, sem outros sintomas neurológicos associados, marcha normal, e sem ataxia. A paciente apresentava níveis de imunoglobulinas IgA, IgG e IgE baixos, com o aumento da classe IgM, linfócitos T normais e níveis de alfa-fetoproteína muito elevados. AT relativamente leve foi suspeitada. Análise genética revelou a presença de duas variantes na sequência codificante do gene ATM. A primeira é uma deleçao de um nucleotídeo na posiçao 3802 (c.3802delG), que resulta na síntese de proteína truncada (p.Val1268Xfs). A segunda variante é uma mutaçao homozigótica missense (c.5948 A>G), que no nível da proteína leva à substituiçao de asparagina (Asn) por serina (Ser) na posiçao 1983 (p.Asn1938Ser). O diagnóstico de síndrome de AT foi confirmado na ausência de ataxia, como uma apresentaçao rara da doença.
Descritores: Síndromes de imunodeficiência, ataxia telangiectasia, ataxia.
INTRODUÇAO
A síndrome de ataxia telangiectasia (AT), também conhecida como síndrome de Louis-Bar, é uma doença neurodegenerativa com envolvimento sistêmico, de herança autossômica recessiva, sendo encontrados portadores heterozigotos em 1-2% da populaçao1. A incidência de AT é de 1 em cada 40.000 a 100.000 nascimentos2. É uma doença heterogênea por envolvimento clínico e genético, caracterizada por ataxia cerebelar, telangiectasias oculocutâneas, infecçoes respiratórias recorrentes, imunodeficiência variável humoral e celular, alta incidência de malignidade, hipersensibilidade à radiaçao ionizante, instabilidade cromossômica e altos níveis de alfa-fetoproteína. Telangiectasias se desenvolvem entre 3-6 anos de idade e têm localizaçao predominantemente na conjuntiva bulbar, e em superfície de orelhas e cotovelos3. Os sintomas neurológicos sao de início precoce, principalmente na forma de ataxia cerebelar. Gatti e cols. relataram que ataxia tipicamente se observava após o início da marcha (12-14 meses), e distúrbios do movimento podem progredir até a necessidade de uma cadeira de rodas (10-11 anos)1,4. Há relatos na literatura de apresentaçao exclusiva de manifestaçoes cutâneas e imunodeficiência combinada sem distúrbios neurológicos5.
Imunoglobulinas A e E sao alteradas, assim como imunoglobulinas G e M. Células T imaturas expressam receptores gama/delta ao invés de receptores alpha/beta, estabelecendo linfopenia progressiva de linfócitos T e B. Linfócitos T CD4+ morrem seletivamente, levando a uma inversao da razao CD4/CD86. Um terço dos pacientes com AT desenvolvem malignidades relacionadas com translocaçoes cromossômicas e quebra excessiva do DNA, o que caracteriza a instabilidade genética da doença, sendo o linfoma a sua forma mais comum7. Os tumores de células T sao 4 a 5 vezes mais frequentes do que os tumores de células B4. Depois de 20 anos de vida, pode aumentar a frequência de tumores epiteliais8.
Ataxia telangiectasia é causada por mutaçoes no gene da Ataxia Telangiectasia Mutada (ATM), que codifica uma proteína do mesmo nome. O papel primário da ATM é a coordenaçao de vias de sinalizaçao intracelular em resposta a quebras no DNA de dupla hélice, stress oxidativo e outros tipos de stress genotóxico. O gene da ATM está localizado no locus de 11q22-239, e codifica uma enzima que está envolvida em etapas de controle do ciclo celular, transporte intracelular e em respostas celulares a danos no DNA, cuja funçao principal é a de proteger o genoma. Sintomas de AT ocorrem como um resultado da perturbaçao na resposta a danos no DNA10.
Manifestaçoes clínicas da síndrome ocorrem inequivocamente na maioria dos pacientes, no entanto, existe um certo grau de variaçao tanto da gravidade como da expressao fenotípica em alguns casos11. Um exemplo disso é o envolvimento neurológico, que apresenta início tardio lentamente progressivo, ou manifestaçoes leves em casos raros12. Folgori e cols. relataram o caso de uma criança de 3 anos com síndrome de AT que apresentou granulomatose exclusivamente cutânea e imunodeficiência combinada grave, sem comprometimento neurológico5. Loeb et cols. relataram dois pacientes com neoplasia linfoide, que receberam tratamento com quimioterapia e apresentaram ataxia, considerada inicalmente secundária à toxicidade do tratamento; posteriormente verificaram que a neoplasia e a ataxia faziam parte da Síndrome AT13. Trimis e cols. descreveram o caso de uma criança de 6 anos com história familiar de câncer de mama, que apresentou telangiectasia ocular e infecçao sinopulmonar sem melhora. Foram realizados estudos imunológicos e genéticos que confirmaram o diagnóstico de síndrome AT sem comprometimento neurológico14. O último caso foi registrado em 2013, de uma paciente de 60 anos que iniciou com ataxia aos 52 anos e apresentava telangiectasias faciais. Tinha história pouco clara de coreoatetose na infância, e ausência de história pessoal ou familiar de infecçao ou câncer15.
O seguinte relato de caso destaca a variabilidade clínica da doença, iniciando com imunodeficiência e telangiectasias oculocutâneas, sem apresentaçao de sintomas neurológicos, particularmente ataxia, e com diagnóstico de síndrome de AT confirmado por estudo genético.
RELATO DO CASO
Paciente do sexo feminino, 8 anos de idade, filha de pais nao consanguíneos, resultado da primeira gravidez a termo, peso normal, saudável ao nascer sem complicaçoes, sem história familiar de doenças relevantes. Apresentou desenvolvimento neurológico normal: sentou-se aos seis meses, rastejando aos oito meses, andou aos 12 meses, palavras dissilábicas aos 18 meses, justapostas aos dois anos, e frases simples aos três anos. A partir de 6 meses de idade passou a apresentar infecçoes de repetiçao: bronquiolite, pneumonia bacteriana, sinusite, infecçoes de ouvido e infecçoes do trato urinário, necessitando de tratamento hospitalar com antibióticos de amplo espectro. Aos 4 anos de idade, procura pela primeira vez o Hospital Universitário Infantil de San José com pneumonia bacteriana associada a componente broncoobstructivo grave, deficiência de crescimento e desnutriçao crônica. Por evoluçao lenta e persistência da infecçao, foi estudada por imunologista pediátrica, que encontrou otorreia fétida bilateral, pneumonia bacteriana, telangiectasia em olhos (Figura 1A) e orelhas, sem comprometimento neurológico. Teve alta com suspeita clínica de síndrome de imunodeficiência primária possivelmente AT, sendo encomendados estudos.
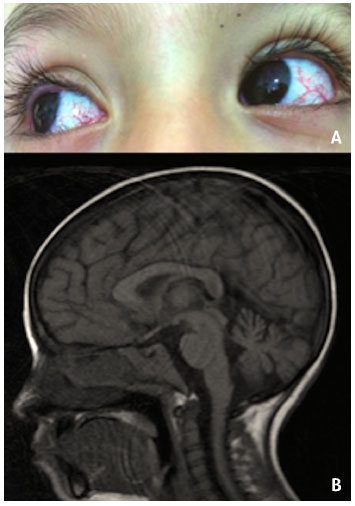
Figura 1 - A) Telangiectasia ocular. B) Ressonância nuclear magnética do cérebro, mostrando atrofia de hemisférios cerebelares e vermis
A tomografia computadorizada de tórax mostrou bronquiectasias císticas comprometendo as vias aéreas periféricas e centrais, e sinais de aprisionamento aéreo. Exames laboratoriais revelaram: eletrólitos no suor 38 mmol/L (valor normal < 40 mmol/L); níveis de imunoglobulinas IgA 0,0 mg/dL (valor de referência VR 58-317 mg/dL); IgG 0,1 mg/dL (VR 805-2421 mg/dL); IgM 4,9 g/L (VR 4.5-25 g/L); IgE nao detectável < 1 UI/mL (VR 1-87 UI/mL). Fenotipagem de linfócitos mostrou: células T CD4+ = 396, CD8+ = 922, CD3+ = 1346, CD4/CD8: 0,43, em níveis normais. Alfa-fetoproteína foi muito elevada, de 89,12 ng/mL (valor de referência até 9 ng/mL).
Desde os 4 anos de vida a criança vem sendo tratada com imunoglobulina humana IV (900 mg/kg/dose) a cada 21 dias. Atualmente em uso de imunoglobulina subcutânea 200 mg/kg semanalmente, com o que apresentou evoluçao satisfatória e diminuiçao significativa das infecçoes. Aos 7 anos, foi estudada por neurologia pediátrica, por alteraçao dos movimentos oculares e cefaleia persistente, com apraxia ocular como único achado neurológico, sem ataxia. Ressonância nuclear magnética (RNM) mostrou atrofia simétrica dos hemisférios cerebelares (Figura 1B).
O estudo genético incluiu cromatografia em que as sequências da paciente foram comparadas com as sequências de tipo selvagem de referência. Duas variantes foram identificadas na sequência codificante do gene ATM. A primeira é uma variaçao heterozigótica em que ocorre deleçao de um nucleotídeo na posiçao 3802 (c.3802delG) (Figura 2A), levando à síntese de proteína truncada (p. Val1268Xfs), já descrita na literatura16. A segunda variante é uma mutaçao missense homozigótica (c. 5948 A> G) nao sinônima, que ao nível da proteína, leva à mudança de uma asparagina (Asn) por uma serina (Ser) na posiçao 1983 (p.Asn1983Ser) (Figura 2B ), já relatada em banco de dados (dbSNP: rs659243)17.
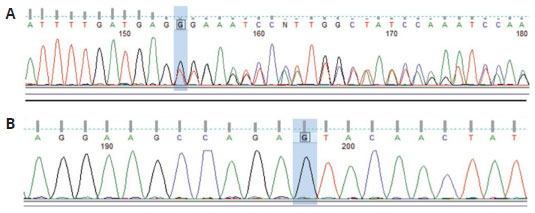
Figura 2 - Cromatografia da sequência codificante do gene ATM. A) Cromatograma indicando a mutaçao heterozigótica c.3802delG. B) Cromatograma indicando mutaçao homozigótica c.5948 A>G
DISCUSSAO
Este caso mostra a variabilidade clínica da síndrome AT; a paciente apresenta-se com imunodeficiência severa e achados clínicos de telangiectasia ao exame ocular, sem comprometimento neurológico. Com o estudo genético, foram encontradas duas variantes identificadas na sequência codificante do gene ATM: c.3802delG (p.Val1268Xfs); e c.5948A>G (p.Asn1938Ser), previamente descritas na literatura. Posteriormente, aos 7 anos de idade, a paciente apresentou apraxia ocular e atrofia cerebral na ressonância magnética, sem ataxia. Heterogeneidade genética e alélica pode ser explicada pela presença de alteraçoes genéticas adicionais. Uma mutaçao conhecida como 5762ins137 esteve associada com retardo da deterioraçao neurológica, sintomas de início tardio, radiossensibilidade intermediária, e menor ou nenhum risco de câncer. Mutaçoes 7271T> G e 8494C> T têm sido associadas ao fenótipo médio e maior expectativa de vida. No entanto, o baixo número de indivíduos e a falta de homozigotos para estas mutaçoes, dificultam correlaçoes estatisticamente significantes 8,11.
O risco de câncer e radiossensibilidade em heterozigotos ATM é quatro vezes maior do que na média da populaçao, particularmente risco de câncer de mama. Os riscos dependem de fatores como tipo de tumor, idade de início do câncer, e se o heterozigoto carrega uma mutaçao non-sense ou uma mutaçao truncada. Estudos epidemiológicos sugerem que os portadores de ATM têm um risco aumentado para doença coronária. Diagnóstico precoce da síndrome AT é importante para a prevençao e tratamento de complicaçoes associadas à imunodeficiência e ao defeito de reparo de DNA, diminuindo riscos de danos cromossômicos e malignidade linforreticular devido à radiaçao ionizante.
AGRADECIMENTOS
Um agradecimento especial aos médicos Drs. Anete Grumach, Ricardo Sorensen, Carlos Restrepo e Alfonso Suárez por suas sugestoes, correçoes e suporte acadêmico.
REFERENCIAS
1. Gatti RA. Ataxia-telangiectasia. In: Scriver CR, Beaudat AL, Sly WS. Metabolic and molecular basis of inherited diseases. 8ª ed. New York: McGraw-Hill; 2001. p. 705-32.
2. Swift M, Morrell D, Cromartie E, Chamberlin AR, Skolnick MH, Bishop DT. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am J Hum Genet. 1986;39:573-83.
3. Muci R, González D, Ravel M. Síndrome de ataxia telangiectasia (enfermedad de Louis Barr). Una rara facomatosis. Gac Méd Caracas. 2013;121:52-6.
4. Sharma A, Buxi G, Yadav R, Kohli A. Ataxia telangiectasia: a report of two cousins and review of literature. Indian J Med Paediatr Oncol. 2011;32:217-22.
5. Folgori L, Scarselli A, Angelino G, Ferrari F, Antoccia A, Chessa L, et al. Cutaneous granulomatosis and combined immunodeficiency revealing Ataxia-Telangiectasia: A case report. Ital J Pediatr. 2010;36:29.
6. Regueiro J, Porras O, Lavin M, Gatti R. Ataxia telangiectasia: a primary immunodeficiency revisited. Immunol Allergy Clin N Am. 2000;20:177-206.
7. Viniou N, Terpos E, Rombos J, Vaiopoulos G, Nodaras K, Stamatopoulos K, et al. Acute myeloid leukemia in a patient with ataxia telangiectasia: a case report and review of literature. Leukemia. 2001;15:1668‑70.
8. Gatti R. Ataxia-Telangiectasia. 1999 Mar 19 [Updated 2010 Mar 11]. In: Pagon RA, Adam MP, Bird TD, et al., eds. Gene Reviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK26468
9. Gatti RA, Berkel I, Boder E, Breedt G, Charmeley P, Concannon P, et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22-23. Nature. 1988;336:577-80.
10. Verhagen MM, Last JI, Hogervorst FB, Smeets DF, Roeleveld N, Verheijen F, et al. Presence of ATM protein and residual kinase activity correlates with the phenotype in ataxia-telangiectasia: A genotype - phenotype study. Hum Mutat. 2012;33:561-71.
11. McConville C, Stankovic T, Byrd P, McGuire G, Yao Q, Lennox G, et al. Mutations associated with variant phenotypes in Ataxia-Telangiectasia. Am J Hum Genet.1996;59:320-30.
12. Taylor A, Byrd P. Molecular pathology of ataxia telangiectasia. J Clin Pathol. 2005;58:1009-15.
13. Loeb M, Lederman H, Winkelstein JA. Lymphoid malignancy as a presenting sign of ataxia-telangiectasia. J Pediatr Hematol Oncol. 2000;22:464-7.
14. Trimis GC, Athanassaki CK, Kanariou MM, Giannoulia-Karantana AA. Unusual absence of neurologic symptoms in a six - year old girl with ataxia - telangiectasia. J Postgrad Med. 2004;50:270-1.
15. Sharrack N, Newrich L, Hadjivassiliou M. A late onset Ataxia with telangiectasia. J Neurol Psychiatric. 2013;84:e2 doi:10.1136/jnnp- 2013-306573.141.
16. Stankovic T, Kidd AM, Sutcliffe A, McGuire GM, Robinson P, Weber P, et al. ATM mutations and phenotypes in ataxia-telangiectasia families in the British isles: expression of mutant ATM and the risk of leukemia, lymphoma and breast cancer. Am J Hum Genet. 1998;62:334-5.
17. dbSNP. Public database of short genetic variations. Disponível em: http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?searchType=adhoc_search&type=rs&rs=rs659243. [Consultado em 10 de março de 2014].
2024 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888